
Gauchers sygdom er en arvelig, såkaldt lysosomal sygdom. Lysosomale sygdomme opstår fordi et eller flere af de enzymer, som nedbryder fedtstoffer og andre molekyler i kroppens celler, enten ikke fungerer ordentligt eller helt mangler.
Ved Gauchers sygdom har kroppen for lidt af enzymet glucocerebrosidase eller mangler helt dette enzym. Glucocerebrosid ophober sig derfor i nogle celler, som man kalder Gaucherceller, disse celler findes primært i milten, leveren og knoglemarven men også i andre væv, f.eks. i lymfesystemet, lungerne, tarmene, og i visse tilfælde i nervesystemet.
Gauchers sygdom er tidligere blevet opdelt i tre typer Type I, Type II og Type III. Der er imidlertid store individuelle forskelle på hvordan sygdommen kommer til udtryk. Det afhænger meget hviken mutiation man har. Derfor går man mere og mere væk fra typeinddelingen og ser mere på hvor alvorlig sygdommen er hos den enkelte person.
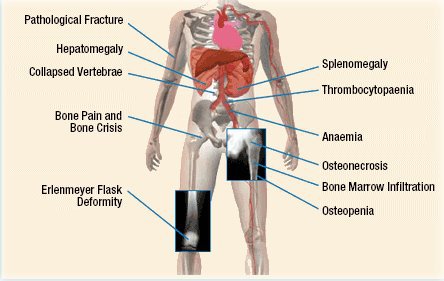
• Type l der er den mest udbredte form viser sig meget forskelligt. Nogle får slet ingen symptomer før sent i livet, andre får lette symptomer og andre igen har symptomer, der kan være invaliderende. Ubehandlet gør aflejringerne at milt og lever kan blive meget store og derfor forårsage blodmangel og reducering af antallet af blodplader. Også skelettet påvirkes og det kan give smerter, på grund af knogleskørhed og deformiteter.
• Type II, viser sig hos helt små børn. Det alvorligste symptom er at hjernen er påvirket, og at børnene ikke lever længere end til 2 års alderen.?
• Type III viser sig i løbet af barnealderen og kan ubehandlet medføre død inden 12-årsalderen. Ud over symptomerne fra Type l, ser man her også vanskeligheder med at flytte blikket til siderne, skelen, gradvist tiltagende demens, epilepsi, balanceproblemer, nedsat hørelse, koncentrasionsbesvær, parkinson, kondinationsproblemer, synkebesvær, langsom reaktion og ufrivillige bevægelser
.
Hvad er symptomerne?
Det er meget forskelligt og afhængigt af typen af Gauchers sygdom, hvilke symptomer den enkelte person får og hvor voldsomme, de er.
Symptomerne på Gauchers sygdom er
• træthed (på grund af ”lav blodprocent”)
• hyppige blødninger og blå mærker, fordi man har for få blodplader
• forstørrelse af milt og lever (stor mave og voldsommen mavesmarter)
• nedbrydning af knoglerne, som kan føre til hyppige brud
• blodmangel (et lavt antal røde blodlegemer, som kan medføre træthed)
• smerter i knoglerne.
Hvordan stiller man diagnosen?
• på baggrund af de symptomer, personen har
• niveauet af enzymaktivitet i en blodprøve
• ved blodprøver kan man se om et andet enzym, chitotrosidase enzymet er forhøjet.
Hvis man udelukkende stiller diagnosen ud fra symptomerne, er der risiko for, at der bliver stillet en fejlagtig diagnose, fordi symptomerne på sygdommen kan variere meget.
Hvem arver sygdommen?
Gauchers sygdom er en såkaldt recessiv arvelig sygdom. Det betyder, at man fra begge forældre skal arve en defekt kopi af det gen, der har betydning for sygdommen. . Har man kun en enkelt defekt kopi af genet (er bærer af genet), får man ingen symptomer, fordi man stadig vil være i stand til at producere nok glucocerebrosidase til at forhindre glucocerebrosid i at ophobe sig.
Kun én ud af 40.000-60.000 i normalbefolkningen har Gauchers sygdom, som forekommer på tværs af racer og etniske grupper. Det er dog den mest almindelige arvelige sygdom blandt jøder af østeuropæisk afstamning (den Ashkenaziske jødiske befolkning). Her har ca. én ud af 40 den arvelige defekt, og ca. én ud af 400 får Gauchers sygdom.
Kan Gauchers sygdom behandles?
Tidligere var behandlingsmulighederne begrænsede. Gaucher patienter fik smerte-behandling, nogle fik fjernet milten, når den blev faretruende stor, eller de fik blodtransfusioner i tilfælde af blodmangel.
I dag kan langt de fleste former for Gauchers sygdom behandles effektivt. Undtaget er dog den alvorlige form, der opstår pludseligt hos spædbørn (type II). Dog kan type 3 heller ikke behandles optimalt. Enzym erstatningen, kan ikke bryde gennem den såkaldte “blod hjer barierer”, så kan ikke behandle det neurologiske og derfor heller ikke centralnervesygstemet. Heldig forsker flere, på at kunne bryde igennem den.??☺️
Standardbehandlingen i dag af Gauchers sygdom består i at tilføre enzymet udefra. Når kroppen får tilført det manglende enzym, bliver glucocerebrosid nedbrudt som normalt. Enzymet bliver tilført via en infusion hver 2. uge enten på hospitalet eller hjemme. For type 1 kan man behandle med en pille, som skal tages 1-2 gange dagligt.